Protein Modelling#
Molecular Dynamics Simulations in Biochemistry#
Individual cellular processes are complex; involving a plethora of reactants, reaction pathways, intermediary states and biological catalysts. Such processes are often very large (\(>1,000,000\) atoms), and with modern computing power, treating such large systems at a full ab initio level is not appropriate. Understanding these reaction pathways in view of developing drugs or treatments for diseases is often an immense undertaking. For example, the structural determination of proteins by experiment requires that these proteins are synthesised, crystallised and purified. One may, however, encounter difficult transfection, suffer from low yields or high losses during purification: all of which lead to very high monetary cost. Crystallisation techniques often do not capture the protein of interest in its active state, and cannot isolate some proteins at all, hence may not be of much benefit to perform.
Computational modelling is generally a useful tool in cases where mechanisms or structures are poorly understood. Additionally when using molecular dynamics, processes can be captured explicitly, thus analysis can be performed in visual and mathematical detail, in an environment in which the protein is active.
Amino Acids and Proteins#
Amino Acids#
Amino acids are the structural units that make up proteins. They join together via dehydration (Fig. 9) to form short polymer chains named peptides, or longer chains called polypeptides or proteins. These polypeditic chains are unbranched, with each amino acid within the chain attached to two neighbouring amino acids. There are a total of 23 amino acids that are naturally incorporated into polypeptides, termed proteinogenic amino acids. Of these, 21 are encoded by the universal genetic code, while the remaining two, selenocysteine and pyrrolysine, are incorporated into polypeptides by unique biological mechanisms.
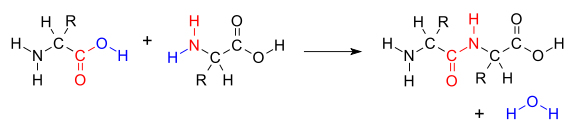
Fig. 9 Dehydration of two amino acids to yield a dipeptide. Each amino acid differs in the R group, thus polypeptide chains have a preserved and regular backbone. Public domain V8rik#
{kind=link}
Proteins#
Proteins are large macromolecules consisting of one or more polypeptide chains. They differ from one another primarily in their sequence of amino acids, which is dictated by the genetic coding responsible for that particular protein. The amino acid sequence results in a folded protein with a specific structure that determines its activity for a particular process. Proteins perform the majority of the vast array of functions required for life within living organisms, including: metabolic catalysis, DNA replication and signal transduction.
Proteins: Form and Function#
The activity and task of a protein, in large part, is determined by its 3D structural characteristics. This is defined by the sequence of amino acids in the polypeptide chains that constitute the protein, which, in turn, is defined by the sequence of genetic code on the encoding mRNA strand.
\(\alpha\)-helices and \(\beta\)-sheets#
Polypeptide chains are not usually linear, in fact, they can adopt very specific conformations which are vitally important to the structural characteristics of proteins. The most common conformations are the \(\alpha\)-helix and \(\beta\)-sheet (Fig. 10), however, there are variants which differ in their symmetry. The \(\alpha\)-helix conformation results when every backbone \(N-H\) group donates a hydrogen bond to the backbone \(C=O\) of the amino acid four residues earlier (\(i+4 \rightarrow i\) hydrogen bonding). Conversely, \(\beta\)-sheets consist of \(\beta\)-strands (linear polypeptides) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted pleated sheet.
There are multiple larger-scale structures in which a number of \(\alpha\)-helical or \(\beta\)-sheets assemble into complicated motifs. These include: coiled-coils, \(\beta\)-hairpin, Greek key motif, \(\beta\)-\(\alpha\)-\(\beta\) motif, \(\beta\)-meander motif and \(\Psi\)-loop motif.
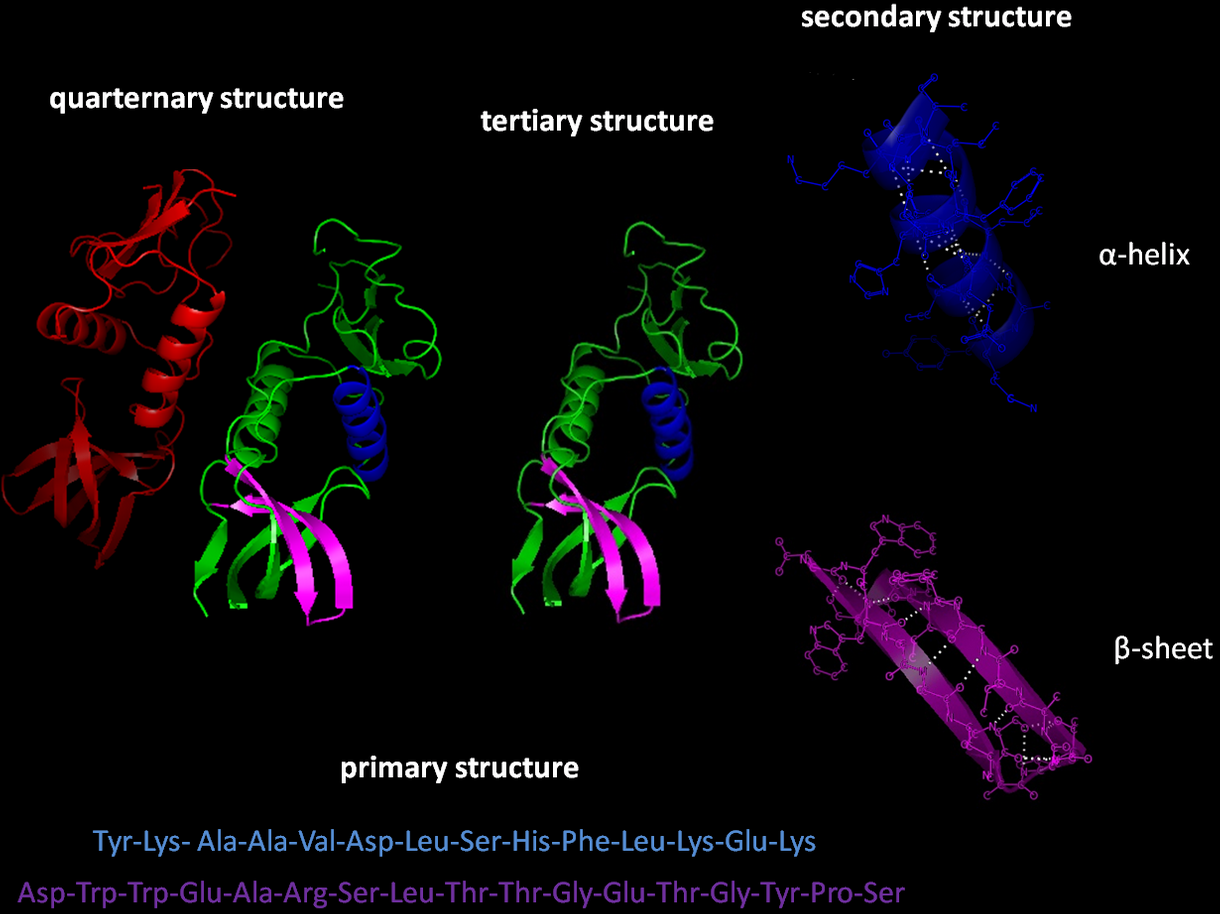
Fig. 10 Hydrogen-bonding is the primary driving force of the formation of \(\alpha\)-helices and \(\beta\)-sheets. This in turn depends on the type and sequence of amino acids involved in the polypeptide. Top alpha helix from the GB1 protein, bottom beta hairpin from the GB1 protein containing a beta sheet connected with a short loop.#
Protein Assemblies#
There are three common structures involving proteins: secondary, tertiary and quaternary structures (Fig. 11). The study of these structures is vital to the understanding of diseases, metabolism and other biological processes.
Secondary Structures#
Secondary structures evolve from the interaction behaviour of hydrogen
bonding formed between the amine hydrogen and carbonyl oxygen atoms
contained in the backbone peptide bonds of the protein. There are a
number of secondary structures observed in nature, the two most common
being the \(\alpha\)-helix and \(\beta\)-sheet described in alpha-helix
Tertiary Structures#
Tertiary structures are proteins which have a single polypeptide chain, with one or more protein secondary structures.
Quaternary Structures#
Quaternary structures involve the folding of multiple folded protein or coiling protein molecules in a multi-subunit complex.
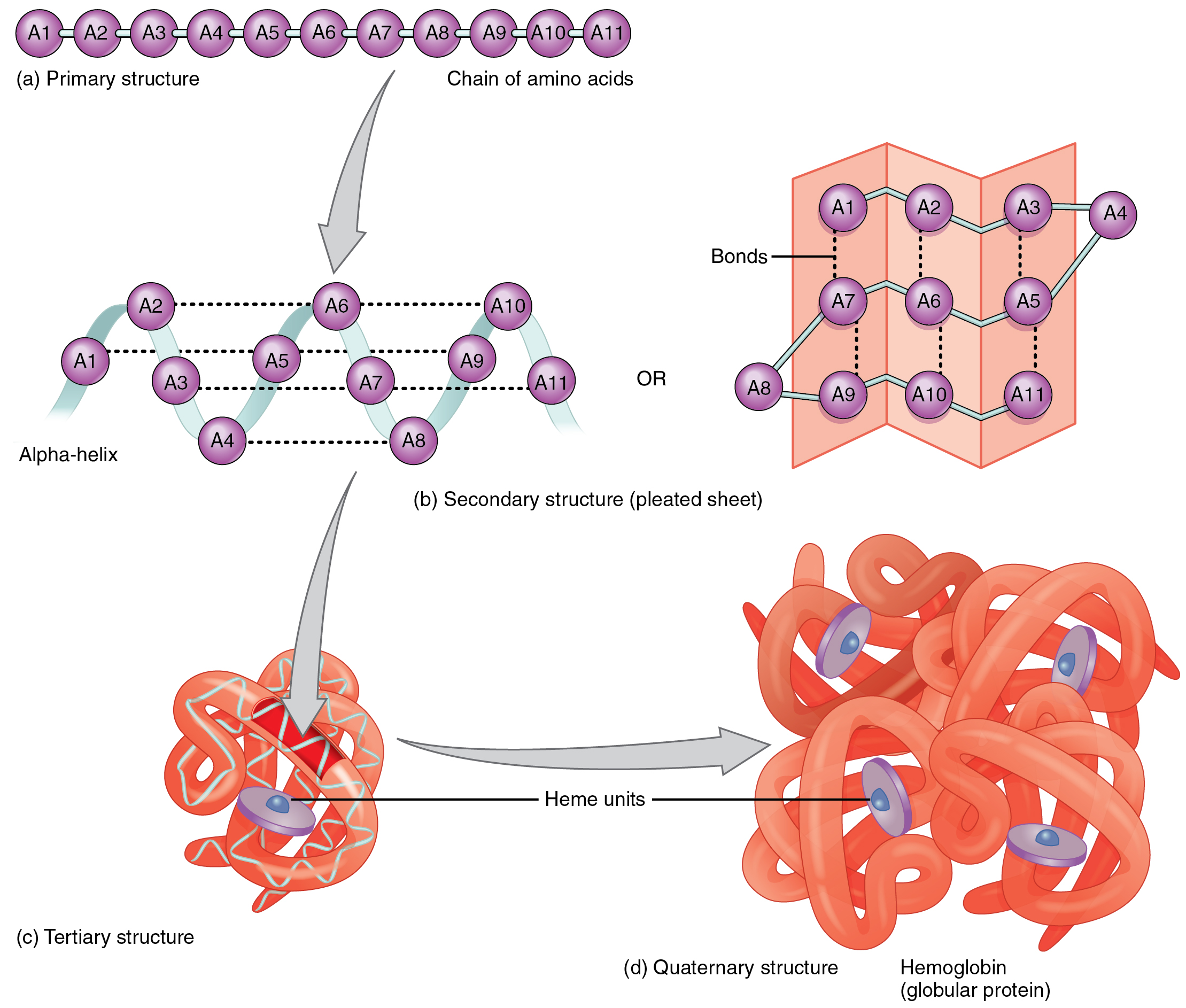
Fig. 11 Depiction of the components of quaternary structures. (CC by SA 3.0 OpenStax College - Anatomy & Physiology,)#
Protein Folding#
Protein folding is the process by which a protein assumes its functional shape or conformation. Each protein exists as an unfolded polypeptide chain when translated from a sequence of mRNA to a linear chain of amino acids (Figure Fig. 12). Early folding already occurs in the exit tunnel of the ribosome (such as formation of alpha helices). After exiting the polypeptide chain then further folds up in a complex process than can involve multiple intermediary state which sometimes require chaperone proteins to fold the protein into the global energy minimum. This free energy minimum exists because displacement of ordered solvent around the hydrophobic core into bulk solvent is favorable (so called hydrophobic collapse, Fig. 12).

Fig. 12 An example of a protein primary chain folding into a complex tertiary structure by hydrophobic collapse. Filled black dots are hydrophobic amino acids. Public Domain Tomixdf#
{kind=link}
Computational methods#
Analysis of the Folding Dynamics of the Trp-cage Miniprotein#
The Trp-cage miniprotein is a designed and stable polypeptide that behaves like larger globular proteins. It is currently the smallest protein to display two state folding properties which is easy to analyze in the laboratory e.g via thermal or chemical denaturation. The small size of this protein makes it an ideal candidate for computational folding simulations, thus it is frequently used to benchmark new computational techniques and as a model system to compare folding morphologies in larger systems. Simulating the folding dynamics of this protein with explicitly defined water molecules is both a lengthy and computationally costly process Fig. 13. Instead, we make use of an implicit solvent model to reduce computational cost and still include, in approximate detail, the effects of water solvation. This is inspired by work of Simmerling et. al.[SSR02] in 2004.
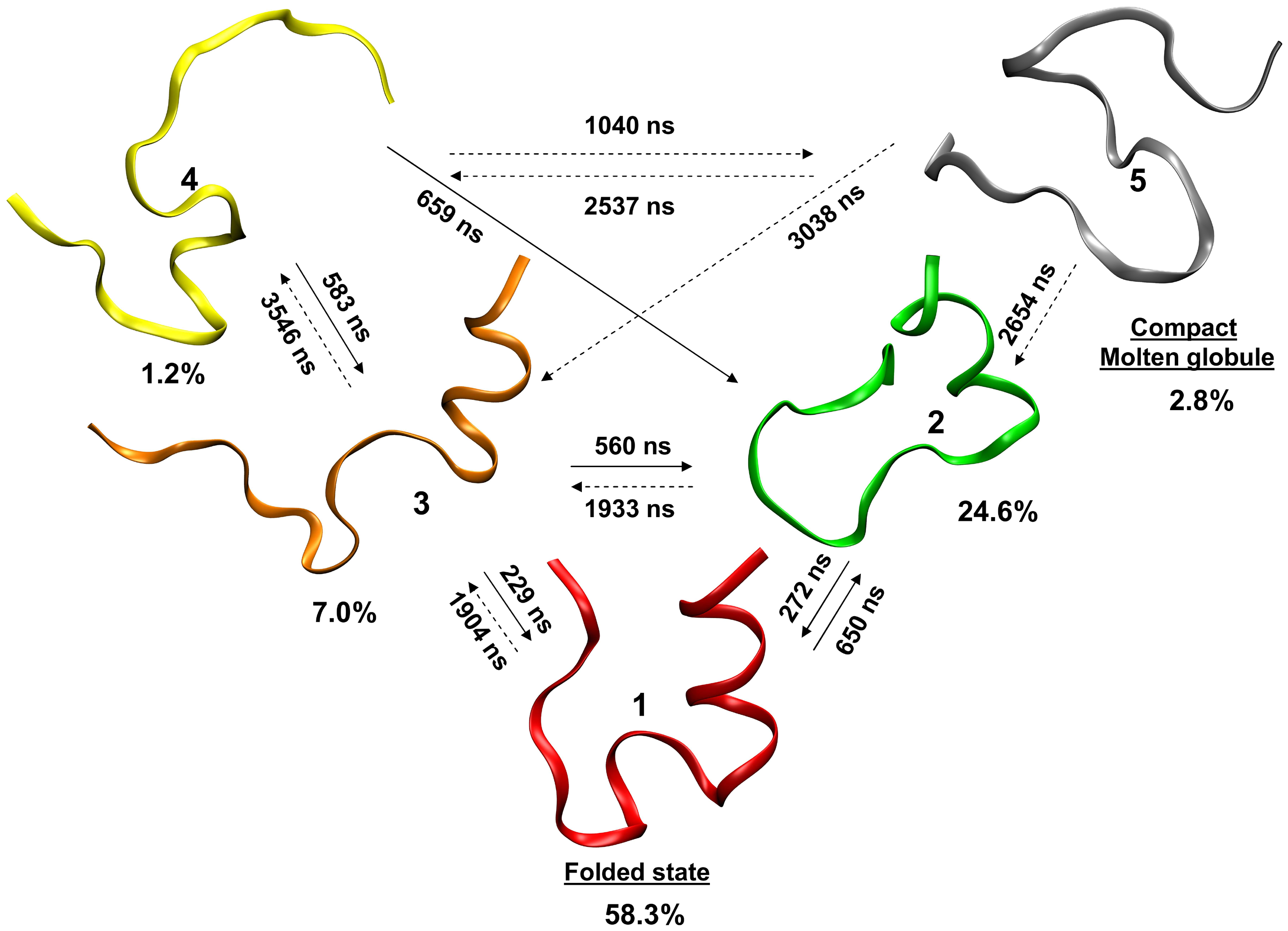
Fig. 13 A computationally determined kinetic model of Trp-cage showing different states in the folding process and their interconversion rates. Yellow unfolded protein, red folded state, gray molten globule (i.e partially folded states) Marinelli et. al. CC by SA#
Fig. 14 Trp-cage starting structure in which cyan represents carbon, red is oxygen, white is hydrogen and blue is nitrogen. Built using the xLeap program within the AmberTools package.#